What is Sickle Cell?
What are the symptoms of sickle cell disease?
Symptoms of sickle cell disease include but are not limited to: abdomen and chest pain, fatigue, breathlessness, delayed growth, jaundice, susceptibility to infections, leg ulcers, swollen extremities, stroke and fever. In addition, symptoms may also include pneumonia, gallstones, swelling of hands and feet, problems during pregnancy, painful erections in men, organ damage, kidney failure and other serious ailments.
What is Sickle Cell Trait?
- 1 in 12 African Americans carry the “silent” Sickle Cell Trait
- 1 in 36 Hispanics carry the “silent” Sickle Cell Trait
Sickle Cell Trait is not Sickle Cell Disease. Sickle Cell Trait (SCT) occurs when one person inherits one sickle cell gene and one normal gene. People with SCT usually do not have any of the symptoms of sickle cell disease (SCD), but they can pass the trait on to their children, that is why education is vital. Knowing if you have SCT can be the difference of giving birth to a child with the disease or not. By knowing your status you can break the sickle cycle. To get tested contact us at 1-844-994-HOPE
How can Sickle Cell be detected?
Detection Methods:
- Screening for sickle cell disease using a simple blood test (ask for a hemoglobin electrophoresis)
- Newborn Screening
- Testing at our office, we provide FREE hemoglobin electrophoresis testing. If you would like to know if you have sickle cell disease, sickle cell trait or any abnormal hemoglobinopathy please schedule an appointment at 1-844-994-Hope.
Is there a Cure?
There is no universal cure for Sickle Cell Disease.
Aggressive medical treatment is necessary. The focus of medical treatment is on prevention of infection, the decrease of symptoms and research. The most commonly used treatment is oxygen, fluids (oral and intravenous), antibiotics, and pain medication. A drug called hydroxyurea, which raises fetal hemoglobin levels, it also reduces painful crises, acute chest syndrome and the need for frequent blood transfusions. However, none of these methods is a cure. In some cases, bone marrow transplant and cord blood transplantation are recommended.
What research is available?
New research, new medications, medical treatments, clinical trials and other research is slowly becoming more available for sickle cell disease patients. For several decades, only hydroxyurea was approved for sickle cell disease. Now Endari, is FDA approved and many others are coming down the pipeline. As certified hemoglobinopathy educators and as an agency member for the SCDAA, we directly aid in research for a potential cure. Please contact our agency and subscribe to our newsletter to learn more about upcoming research and developments regarding sickle cell disease and sickle cell trait.
If you would like to learn about upcoming and current clinical trials for sickle cell disease click here. For upcoming trials for sickle cell trait click here.
What is Sickle Cell Disease?
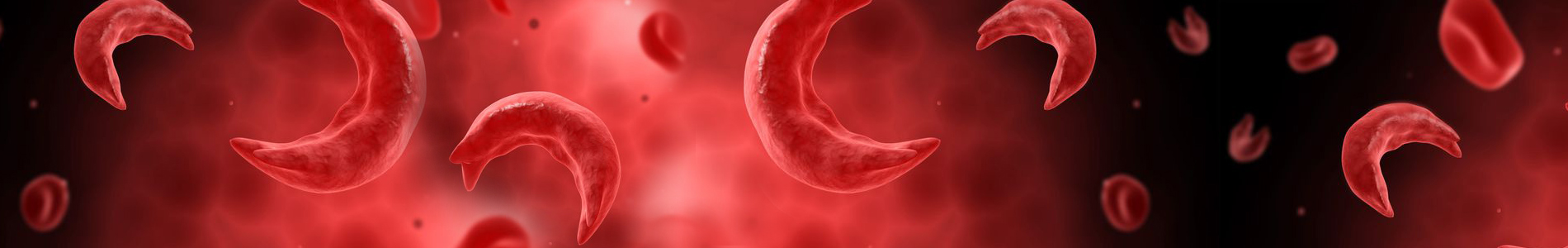
Sickle cell disease is an inherited life-threatening blood disorder that affects the red blood cells. These sickled cells carry less oxygen to parts of the body and when these misshaped cells are clumped together causes pain crisis. It affects African Americans, Hispanics, Greeks, Italians, East Indians, Saudi Arabians, Asians, Syrians, Turks, Cypriots, Sicilians, Caucasians, and others. Sickle cell disease is quietly devastating. Its symptoms can occur in any part of the body. Persons with the disease extremely vulnerable to infections. They may also suffer from jaundice deterioration of joints, kidney infections, recurrent severe pain episodes, strokes, blindness and a shortened life expectancy. Sickle cell disease can lead to other ailments. These include strokes, kidney and liver problems. There is no cure.
Specifically, Sickle cell disease is characterized by defective hemoglobin (a protein in red blood cells that carries oxygen to the tissues of the body).
Sickle cell disease inhibits the ability of hemogloblin in red blood cells to carry oxygen. Cells containing normal hemoglobin are smooth, disk-shaped, and flexible, like doughnuts without holes, so they can move through the vessels in our bodies easily. Cells containing sickle cell hemoglobin are stiff and sticky and form into the shape of a sickle or crescent, like the letter C, when they lose their oxygen. These sickle cells tend to cluster together, and cannot easily move through the blood vessels. The cluster causes a blockage in small arteries or capillaries and stops the movement of healthy, normal oxygen-carrying blood. This blockage is what causes the painful and damaging complications of sickle cell disease.
Sickle cells only live for about 10 to 20 days, while normal red blood cells can live up to 120 days. Also, sickle cells risk being destroyed by the spleen because of their shape and stiffness. The spleen is an organ that helps filter the blood of infections. Sickled cells get stuck in this filter and die. Due to the decreased number of healthy red blood cells circulating in the body, a person with sickle cell disease is chronically anemic. The spleen also suffers damage from the sickled cells blocking healthy oxygen carrying cells and typically infarcts in the first few years of life. Without a normal functioning spleen, these individuals are more at risk for infections. Infants and young children are at risk for life-threatening infections.
Resources For You
Below is a list of programs and important resources that can help you.